
Clinical Consequences of Uremia
Read
3. Clinical Consequences of Uremia
Gastrointestinal Consequences
Gastrointestinal intoxications including anorexia, nausea, vomiting, fetid breath, stomatitis, oral ulcerations colitis, diarrhea, intussusception and ileus are the most common and prominent clinical signs of uremia. These lesions and dysfunctions act singularly or in concert to induce gastrointestinal pathology.
Excess urea secreted in salivary and gastric juices is converted by urease producing bacteria to ammonia that directly damages the mucosa. Uremic toxins also injure the gastric mucus, mucosa, submucosa or vasculature, thereby reducing the protection afforded by the gastric mucosal barrier. Reduced renal clearance of gastrin leads to hypergastrinemia and stimulation of gastric acid production.
Increased diffusion of acid into the gastric wall induces inflammation, ulceration, and hemorrhage, in addition to perpetuating uremic toxin induced gastric injury. Vomiting occurs secondary to gastritis in addition to the direct effect of uremic toxins on the chemoreceptor trigger zone.
Figure 5. Oral lesions in uremic stomatitis/gingivitis. (© DJ Chew).

Neuromuscular Consequences
The two major neurologic complications of uremia are uremic encephalopathy and neuropathy. Uremic encephalopathy is a term that reflects diffuse and non specific alterations of the cerebral cortex. The severity and progression of the neurological signs is generally correlated with the magnitude and progression of the azotemia. Typical signs include a progressive decline in alertness and awareness, dullness, lethargy, impaired mentation, altered behavior, confusion, stupor, tremors, ataxia, muscular cramps, fatigue, muscle weakness, seizures and coma. The neurological signs are due to the effects of uremic toxins, hyperparathyroidism, hypocalcemia, hypokalemia and hypertension.
The Doppler technique and oscillometry are very common methods for detecting hypertension. Doppler is the recommended technique for cats. The oscillometric measurements in dogs can be unreliable due to differences in conformation, obesity or a thick coat (Stepien, 2001). The animal’s adaptation to the environment is critical to the interpretation of arterial pressure measurements, as stress can induce erroneous results. Six to ten measurements are recommended. (© J-C Meauxsoone).
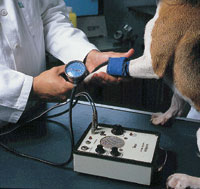
The Doppler technique and oscillome try are very common methods for detecting hypertension. Doppler is the recommended technique for cats. The oscillometric measurements in dogs can be unreliable due to differences in conformation, obesity or a thick coat (Stepien, 2001). The animal's adaptation to the environment is critical to the interpretation of arterial pressure measurements, as stress can induce erroneous results. Six to ten measurements are recommended.
Cardiopulmonary Consequences
Cardiopulmonary complications include hypertension, uremic cardiomyopathy, uremic pericarditis, pulmonary edema and uremic pneumonitis. Abnormalities in fluid, electrolyte and acid base balance may contribute to alterations in cardiac contractility and excitability. Azotemia and overhydration play a role in pericarditis, uremic cardiomyopathy and pulmonary edema. Hypertension arises secondary to a combination of activation of the renin-angiotensin aldosterone system, sodium retention, plasma volume expansion, activation of the sympathetic nervous system, decreased activity of vasodilatory substances, increased cardiac output, increased total peripheral vascular resistance and secondary hyperparathyroidism. Systemic hypertension predominately targets the kidneys (glomerulosclerosis), heart (left ventricular hypertrophy, myocardial ischemia), eyes (retinal detachment, hyphema, retinal hemorrhage) and brain (hypertensive encephalopathy, dementia, cerebrovascular hemorrhage). Uremic pneumonitis refers to the formation of a high protein pulmonary edema which is presumably a consequence of uremic toxins damaging alveoli and increasing capillary permeability.
Ocular Consequences
Common manifestations of advanced uremia include scleral and conjunctival injection and ocular pathology secondary to systemic hypertension. Opthalmoscopic findings include reduced pupillary light reflexes, papilledema, retinal arterial tortuosity, retinal hemorrhage, retinal detachment, hyphema, anterior uveitis and glaucoma. Ischemia and retinal degeneration result from sustained retinal arteriolar vasoconstriction which is an attempt to auto-regulate local blood flow in the face of sustained chronic hypertension.
Metabolic and Endocrine Consequences
The kidney is responsible for the degradation of many peptide hormones and loss of this catabolic function can result in metabolic derangements caused by hormone excess. Deranged insulin metabolism may also contribute to hyperlipidemia. Other hormonal alterations include increased concentrations of gastrin, glucagon, growth hormone, prolactin and luteinizing hormone. Serum T4 concentrations are low and there is impairment in the conversion of T4 to T3 (euthyroid sick syndrome).
Fluid, Electrolyte and Acid-base Consequences
Metabolic acidosis is a frequent finding in renal disease and results primarily from the inability of the kidney to excrete hydrogen ions and regenerate bicarbonate. Chronic acidosis causes progressive bone demineralization, urinary calcium loss, hypokalemia and increases skeletal muscle protein catabolism which exacerbates azotemia.
Hyperphosphatemia is one of the most common regulatory derangements of CRF that arises secondary to reduced glomerular filtration of phosphorus. Hyperphosphatemia contributes to renal secondary hyperparathyroidism (see below), reduced calcitriol levels, soft tissue calcification, renal osteodystrophy and hypocalcemia. Soft tissue mineralization develops as the calcium x phosphate product exceeds 60 (concentrations are expressed in mg/dL).The most commonly targeted organs include the gastric mucosa, bronchial walls, myocardium, endocardium, renal interstitium, glomeruli, lungs, and intercostal muscles. Renal mineralization will promote interstitial inflammation, fibrosis and progression of renal failure.
Hypokalemia is a common abnormality associated with chronic renal failure. The mechanism is unclear and includes excessive urinary potassium loss, inadequate dietary potassium intake, and acidifying diets. Hypokalemia causes generalized muscle weakness and pain which may present as cervical ventroflexion and a stiff, stilted gait. Hypokalemia also impairs protein synthesis, promotes weight loss, a poor hair coat and contributes to polyuria by decreasing the renal responsiveness to ADH. Chronic potassium depletion may indeed impair renal function by inducing a reversible, functional decline in GFR in addition to promoting renal injury by enhancing ammoniagenesis.
Renal Secondary Hyperparathyroidism
Renal secondary hyperparathyroidism is a clinical syndrome that is characterized by increased secretion of parathyroid hormone (PTH). PTH secretion is stimulated by hypocalcemia and decreased plasma calcitriol concentrations. Hypocalcemia is a mass action (i.e. calcium x phosphate remains constant) consequence of renal retention of phosphate.
Calcitriol production is regulated at the level of the 1-α-hydroxylase enzyme in the kidney. Phosphate in excess and loss of functional renal mass result in a decrease in 1-α-hydroxylase activity and reduces the conversion of 25-dihydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (calcitriol). Calcitriol deficiency reduces the intestinal absorption of calcium, reduces the release of calcium and phosphate from bone, reduces renal reabsorption of calcium and phosphate and increases the synthesis and release of PTH (Figure 6aand Figure 6b).
Figure 6a. Influence of PTH on the metabolism of calcium and of phosphorus.

Figure 6b. Mechanism of development of renal secondary hyperthyroidism in CRF.

Initially the increased PTH concentration activates the remaining 1-alpha hydroxylase enzyme with a compensatory increased in calcitriol concentrations. However, with disease progression stimulation of 1-α-hydroxylase becomes ineffectual and calcitriol concentrations remain low. Complications of renal secondary hyperparathyroidism include osteodystrophy, soft tissue calcification, skeletal decalcification, cystic bone lesions, bone pain and growth retardation. Osteodystrophy most commonly occurs in immature patients and is recognized by demineralization of the bones. The teeth become movable and the jaw can be bent or twisted without fracturing (rubber jaw). Facial distortion may occur secondary to connective tissue proliferation. PTH has also been implicated as a uremic toxin and may contribute to the progression of renal failure.
Hematologic Consequences
Normocytic normochromic non-regenerative anemia is the most common abnormality in uremia. The pathogenesis is multifactorial and includes inadequate production of erythropoietin by the diseased kidneys, reduced life span of red cells, nutritional deficiencies, uremic toxin induced inhibition of erythropoiesis and blood loss with consequent iron deficiency. Anemia will contribute to the clinical signs of lethargy and inappetance. Neutrophil function and cell mediated immunity is impaired in uremia, predisposing the uremic patient to infection. The specific causes of renal failure associated immunocompromise is not completely understood, although malnutrition, uremic toxins, PTH and vitamin D concentrations may be involved.
The hydration state is evaluated by clinical examination, the measurement of hematocrit and total plasma proteins. (© J-C Meauxsoone).

1. Adams LG. Phosphorus, protein and kidney disease. Proceeding of the Petfood Forum 1995 (13-26).
2. Bauer JE, Markwell PJ, Rawlings JM et al. Effects of dietary fat and polyunsaturated fatty acids in dogs with naturally developing chronic renal failure. J Am Vet Med Assoc 1999; 215: 1588-1591.
About
How to reference this publication (Harvard system)?
Affiliation of the authors at the time of publication
1Royal Canin USA, MO, USA. 2Experimental Physiopathology and Toxicology, National Veterinary School of Toulouse, Toulouse, France.


Comments (0)
Ask the author
0 comments