
Developing in vivo/in vitro Correlations for Parenteral Products
J. Shah
Read
Introduction
Correlation between an in vitro performance of formulation to its in vivo performance is beneficial for a number of reasons such as the following [1-3]:
- IVIVC may reduce the number of clinical studies needed
- IVIVC may support request for waiver of BE studies if a clinical study has been conducted on the highest strength formulation
- Waiver of BE studies for Level-3 changes based on SUPAC-MR guidance
- Allows sponsor to set wider and clinically relevant specifications for release
- Support atypical conditions used for the in vitro release method
Due to the above stated benefits of in vitro-in vivo correlation (IVIVC), it has been successfully used and demonstrated for oral products, however, IVIVC for parenteral modified release products is challenging for reasons described below [13]. Modified release parenteral products consists of liposome products such as Ambisome and Depocyt that are intended for targeting to a specific tissue or an organ and sustained release products such as Zoladex and Lupron Depot intended for sustained plasma levels [2]. For parenteral products intended for targeting, plasma levels may not be as meaningful as local tissue or organ levels for efficacy and thus IVIVC based on plasma levels is not appropriate. However for such products, biomarkers, tissue levels or surrogate efficacy end-points may be monitored for correlation with in vitro performance and to establish an in vitro-pharmacodynamic correlation.
Parenteral sustained release (SR) formulations, conventionally known as depots, in contrast to oral controlled release formulations, are generally designed to maintain plasma levels for more than a few weeks, preferably for a month or longer [4,5]. These formulations are usually administered by either a subcutaneous (SC) or an intramuscular (IM) injection, which provides sustained plasma levels for weeks to months. Parenteral sustained release is achieved using a variety of technologies such as microsphere, in situ gels, implants, lipophilic prodrugs delivered in oil and aqueous suspensions of poorly soluble drug form (Table 1 and Fig. 1).

Table 1. Representative commercially available parenteral SR products.
Figure 1. How is release controlled from parenteral SR formulations?

The release mechanism for the various SR technologies is very diverse ranging from diffusion and erosion for biodegradable polymer based microspheres and gels to dissolution controlled release for aqueous and oil based suspensions. Biodegradable polymer based microspheres and in situ gelling depots have been used more frequently recently to provide SR following IM and SC administration [5]. However, a large burst is often observed when water soluble drugs are delivered from these microspheres and gelling depot formulations. The release duration for the parenteral SR products range from a few weeks (Risperdal Consta, 2 weeks), months (Depo Provera, 3 month) to a year for an implant such as Norplant. Due to the different therapeutic indications and durations of release, the total dose as seen in Table 1 range from a few milligrams to 150 mg. The above diversity of parenteral SR products makes it difficult to develop a predictive in vitro release method and an IVIVC for them challenging for the following reasons:
- Different pharmacodynamic rationale for modified release parenteral products such as targeting versus extended release application and hence plasma levels may not be meaningful for targeting applications. In such application, suggestion would be to use biomarkers, tissue levels or surrogate efficacy end-points for targeted delivery.
- Different routes of administration such as SC versus IM and thus significant physiological differences such as the amount of fluid present, blood flow rate and the injection volume that can be used.
- Different durations of release as in the following examples, and thus it is questionable if a practical real time in vitro release method can be developed for example for a 3 month formulation.
- Risperdal Consta, 2 week
- Lupron Depot, 1 month
- Depot Provera, 3 month
- Norplant, 1 year
- Different mechanisms of release for various SR parenteral formulations such as bioerosion and diffusion controlled release for PLGA based formulations versus intrinsic dissolution controlled release for suspensions. Therefore, it is questionable if a general broadly applicable release method can be developed that enables the release mechanism to be retained during the study so that a discriminatory and predictive release profile obtained for a variety of parenteral SR products.
- Burst release is generally observed with many parenteral SR formulations which is difficult to model and predict.
Considerations in Development of a Predictive In Vitro Release Method
Ideally, the in vitro release method should simulate the in vivo physiologic conditions for the intended site and route of administration with the following variables to consider:
- Use release medium with pH, buffer capacity and ionic strength simulating physiological environment at the administration site at the physiological temperature.
- Consider use of relevant enzymes if they may play a role in biodegradation and erosion mediated release of the drug from the formulation.
- The release studies should be conducted with realistic injection volumes. For example, a release profile obtained with 0.1 ml injection volume may not predict in vivo release of 1 ml dose particularly for a depot technology based on diffusion-controlled release that is dependent on the surface area/volume ratio of the depot at the injection site.
- If sink conditions do not exist in vivo for the drug when administered as a suspension, it may be appropriate to conduct in vitro release under non-sink conditions as well.
- Other physiological factors such as protein binding, fluid dynamics, fibrous encapsulation and in vivo biodegradation and erosion may play a significant role in in vivo release from parenteral sustained release formulations, however they are difficult to simulate in an in vitro release method.
In addition to simulating physiological conditions, release method should be robust and in vitro release profile should be independent of method variables. If the delivery system or the formulation controls the release, release across the aqueous hydrodynamic diffusion boundary layer should not be rate limiting. Therefore, an attempt should be made to demonstrate that the in vitro release is independent of the fluid dynamics created by different agitation rate or flow rate in a continuous flow release method, and formulation solely controls the release. An in vitro release method should be able to support the proposed mechanism for release from the formulation. For example, for a dissolution-controlled formulation such as suspension, release method should be able to demonstrate the anticipated effect of particle size on release. Similarly, the burst should be reduced from a PLGA based in situ gel or microsphere when the molecular weight of PLGA used is increased. A release method that results in a reproducible release profile that is method variables independent, and the release kinetics supportive of release mechanism will likely be discriminatory for formulation variables. Such a method will differentiate between acceptable and unacceptable formulations and at the least suitable as a good quality control test.
Developing an In Vitro Release Method as a QC Test for Parenteral SR Formulations
Currently there are no suitable USP dissolution/release apparatus for parenteral SR formulations since USP dissolution apparatuses have been developed primarily for immediate and modified release oral dosage forms [3]. The methods that can be considered for parenteral SR formulations and often cited in the literature are the following [2]:
- Flow through method in which the release medium is circulated while the formulation is retained in an apparatus like USP Dissolution apparatus 4. The fluid dynamics in this method may closely simulate blood flow in the muscle for IM depots.
- Dialysis bag method in which formulation is placed in a semipermeable membrane placed in a release medium that is stirred continuously. The commercially available dialysis bags ensure physical separation of the formulation from the release medium yet allowing rapid equilibrium across the membrane for small sized molecules. However, the diffusion boundary layer effects for the dialysis membrane may underestimate the potential burst and attempts must be made to evaluate it.
- In case of a formulation that is a solid as an implant or that forms a semi-solid gel in vivo, dissolution in a vial with periodic sampling or with complete replacement of release medium at each time point can be used. However in this method, it can be difficult to maintain physical separation of formulation from the release medium and this may underestimate the total amount released.
- Implant retrieval following SC injection in a suitable pre-clinical model and estimating drug remaining in the implant has been used to estimate in vivo release for SC implants and in situ gel forming parenteral SR formulations [5]. However, clearly this method is far more resource intensive and not very suitable as a QC test.
As mentioned earlier, release method should be discriminatory, however test conditions that are likely to be discriminating are not obvious. The discriminating power of the test is some times difficult to demonstrate particularly if a solubilizer is used to maintain sink conditions since use of solubilizer swamps any differences in formulations that the method is expected to differentiate. In contrast, if the release is conducted under non-sink conditions, incomplete release is observed. At early stages of product development, aberrant formulations are not typically available to demonstrate discrimination, and thus it is not obvious how to set meaningful specifications without sufficient preclinical and clinical pharmacokinetic data. The development of an in vitro release method for veterinary products is deemed to be more challenging and difficult due to the larger size of the delivery system, large doses for longer durations and for implants, unique size and shapes of implant [6].
Developing In Vitro-In Vivo Correlation (IVIVC)
Due to the reasons outlined above, IVIVC for parenteral formulations is challenging and has not been demonstrated successfully for as many products as modified release oral products. However an attempt has to be made due to the benefits outlined above and also to rationally develop a product and set the appropriate specifications. At the early stages, while a release method is being developed a suitable preclinical model should be used as well to assess the effect of formulation variables on pharmacokinetic profile. This provides an opportunity to correlate in vitro release with the in vivo performance of the formulations in preclinical models. Since very few formulations can be tested clinically, even IVIVC with preclinical data can be used to assess the predictive ability of the in vitro release method. When the clinical data is available even for one formulation, IVIVC should be explored and any type of IVIVC from a point-to-point (Type A) to a single point correlation (Type C) may be useful [1,7]. Furthermore correlation between preclinical and clinical pharmacokinetics should be evaluated since if preclinical pharmacokinetics is predictive of clinical pharmacokinetics, in vitro release method and preclinical pharmacokinetics studies may be sufficient to develop and optimize the formulation.
For Parenteral SR formulations for long durations, it may not be practical to develop an in vitro release method in which case, an accelerated in vitro release method may be developed, provided mechanism of release does not change. Few of the commonly used approaches to accelerate release are use of higher temperature or non-physiological pH to accelerate biodegradation. However it is important to not change the characteristics of the delivery system such as solid-state nature of the PLGA by using a temperature higher than its glass transition temperature. Alternatively, time scaling can be used to develop a correlation between de-convoluted in vivo release profile and in vitro release [7]. Another option might be to use the Levy plot in which, times for a fixed percentage of drug to be release in vitro and in vivo are correlated [8].
Case Studies on IVIVC for Parenteral SR Formulations
At the conference, case studies demonstrating successful IVIVC in preclinical models for parenteral SR formulation from literature will be presented (see Attached Slides). Two case studies on delivery of antibiotics, cefazolin and vancomycin from implants for prevention of post-surgical wound infection demonstrate the principles described above [9-11]. An in vitro release method was developed for cefazolin and vancomycin that confirmed the anticipated mechanism of release; The release of cefazolin and vancomycin from the lipid based insoluble matrix followed the square root of time release kinetics expected for the release of a small water soluble molecule from an insoluble matrix. The release profile was independent of method variables and it correlated well with a physiologically relevant release method in which the matrix was implanted in an agar gel at physiological temperature simulating the intended mode of use for the delivery system. The resulting release profile demonstrated type A, IVIVC for both cefazolin and vancomycin implants with a preclinical pharmacokinetic model [9-11].
Negrin et al. (2004) recently demonstrated type A, IVIVC for PLGA based implants of methadone of 1-week and one month durations [12]. In case of release with a lag-time that is often observed for PLGA based delivery systems, Schliecker et al. demonstrated the use of Levy plot to get IVIVC for a one month buserelin implant [8].
Acknowledgements
Saleh Allababidi and Dakshina Murthy Chilukuri for their thesis work on cefazolin and vancomycin implants. Parenteral Drug Association Foundation, Osteotech Inc. and Medical University of South Carolina for funding the cefazolin and vancomycin research work presented. I would also like to thank Pfizer Global Research & Development for support of the work on this manuscript.
Figure 2. Case Study 1: SC site specific delivery of cefazolin with implants release of 10% cefazolin from implant formulation (n=3).

Figure 3. Release kinetics does support the release mechanism and effect of formulation variables predictabe.
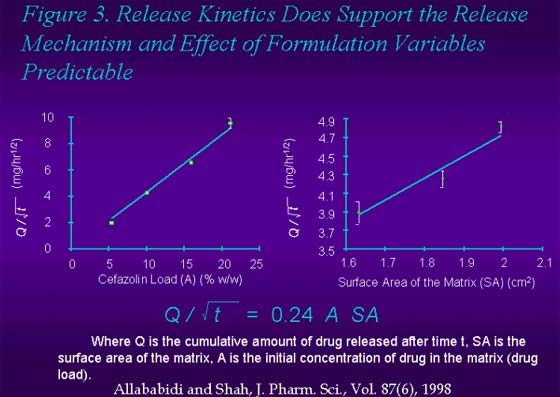
Figure 4. Release kinetics in physiologically relevant conditions.

Figure 5. Spatial release of cefazolin into the Agar gel. Different zones represent area surrounding the implant.

Figure 6. Comparison of release in the gel and the vial method: indicates similar release kinetics.

Figure 7. Extending the release duration by dry coating.

Figure 8. Pharmacokinetics in rat (n=6) of cefazolin delivered with implants.

Figure 9. In vitro versus in vitro release kinetics for cefazolin.

Figure 10. Pharmacokinetics in rat (n=6) of vancomycin delivered with implants (similar formulation and mechanism to cefazolin implants).

Figure 11. Comparison of in vivo release of vancomycin from implants in male sprague-dawley rats with in vitro release conducted in pH 7.4 PBS at 37°C.
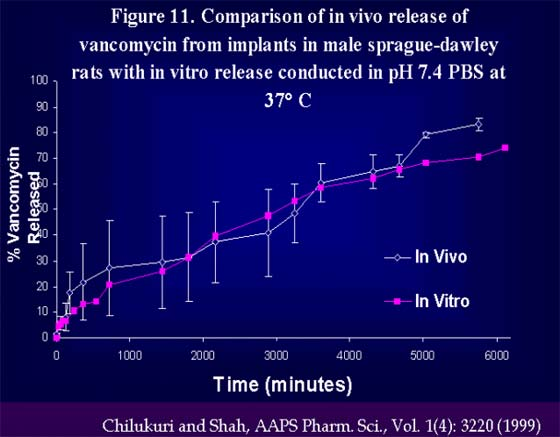
Figure 12. Level A type IVIVC. Correlation of in vitro-in vivo release of vancomycin from GMS implants.

Figure 13. In vitro release and pharmacokinetics profile of methadone implants.

Figure 14. IVIVC for 1-month methadone implant.
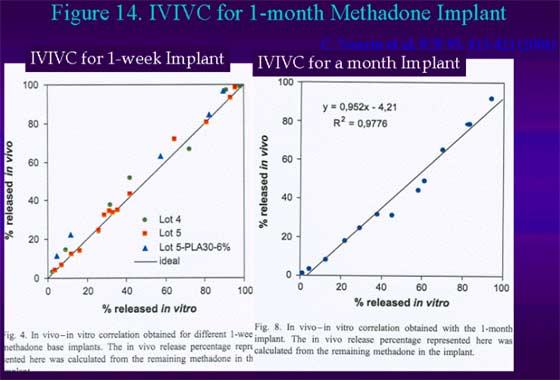
Figure 15. IVIVC of buserelin from PLGA implants.


Comments (0)
Ask the author
0 comments